TOPICS
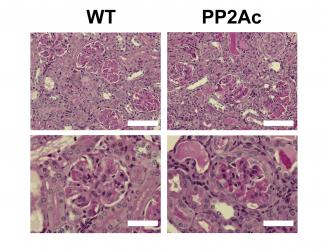
Role of protein phosphatase 2A as a regulator of inflammation
Early studies
reported that the catalytic subunit of the serine/threonine phosphatase PP2A
(PP2Ac) is overexpressed by T cells from patients with systemic lupus
erythematosus (SLE). In order to investigate a possible pathogenic role of such
defect, we generated a transgenic mouse that overexpresses PP2Ac in T cells.
This mouse exhibited an abnormal susceptibility to the development of immune
complex-mediated glomerulonephritis (CrispĂn et
al., J Immunol 2012). In
further studies, we demonstrated that the phenotype was caused by epigenetic
modifications that facilitated the transcription of pro-inflammatory genes
(e.g. Il17a) in an IRF4-dependent
manner (Apostolidis et al., J Biol Chem 2013). PP2A is also an essential element in the
biology of regulatory T cells because its conditional deletion in FoxP3+
cells abolishes their suppressive capacity and triggers a severe systemic
autoimmune disease (Apostolidis et al.,
Nat Immunol 2016).
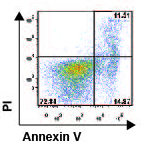
Mechanisms that control the length of immune responses
In order to better
understand the biology of PP2A and its contributions to the development of
autoimmunity, we analyzed the expression of its regulatory subunits in human T
cells. We found that B55Ăź is induced in activated T cells upon cytokine withdrawal and its
presence is necessary and sufficient for the T cells to undergo apoptosis
induced by IL-2 withdrawal (CrispĂn et al., Proc Natl Acad Sci USA 2011). By
analyzing the kinetics of apoptosis and B55Ăź expression in T cells from
patients with SLE, we identified a group of patients whose T cells are
resistant to the induction of apoptosis during cytokine withdrawal.
Importantly, IL-2 deprivation did not induce B55Ăź upregulation in these
patients suggesting that abnormalities in its expression may contribute to
autoimmunity by allowing the survival of activated T cell clones.
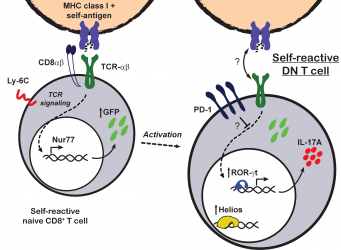
CD8 downregulation as a mechanism of peripheral tolerance
Patients and mice with lupus frequently have increased numbers of TCR-αß+ double negative (DN) T cells. DN T cells are a normally scarce T cell subpopulation that lacks the expression of the CD4 and CD8 coreceptors. In initial studies, we reported that DN T cells can produce pro-inflammatory cytokines (i.e. IL-17 and IFN-gamma) and infiltrate target organs of patients with SLE (CrispĂn et al., J Immunol 2008). Later, we reported that DN T cells are generated by loss of CD8 that occurs after the activation of CD8+ T cells (CrispĂn et al., J Immunol 2009). In order to understand the factors that govern CD8 to DN conversion, we used an in vivo adoptive transfer model. We learned that when CD8+ T cells are productively activated, as in the context of an infection, expression of CD8 is maintained. However, when CD8+ T cells encounter cognate antigen presented as self, they undergo an inactivation process that includes the downregulation of CD8 and the expression of high levels of inhibitory molecules including PD-1 and the transcription factor Helios (RodrĂguez-RodrĂguez et al.,

Biological effects of risk alleles associated to autoimmune diseases
Genome-wide
association studies (GWAS) have identified a relatively large number of genes
and loci associated to SLE and other autoimmune diseases. These analyses detect
common single nucleotide polymorphisms (SNP) whose frequency is significantly higher
in patients than in healthy controls. One of the SNPs that is thought to confer
a higher risk for the development of SLE is located in ITGAM, the gene that encodes the alpha chain of the adhesion
molecule Mac-1. The risk allele of ITGAM
represents a non-synonymous mutation that alters the protein (R77H). In order
to determine the effects of the R77H substitution, we generated cells lines
expressing the wild type or the risk variant of Mac-1. In vitro studies
revealed that the R77H variant diminishes the ligand-binding capacity of Mac-1,
in particular under shear flow (Rosetti
et al, J Immunol 2012). Further studies demonstrated that the risk
allele is unable to form “catch bonds”, a molecular mechanism that allows
integrins to bind with high affinity (Rosetti et al, Cell Reports 2015). To evaluate
the in vivo consequences of decreased Mac-1 function, we analyzed mice that
lack Mac-1 and express the human FcgRIIA. Intravenous injection of immune complex
rich sera obtained from patients with SLE caused the glomerular deposit of
large amounts of immune complexes. Strikingly, absence of Mac-1 was associated
to a dense infiltration of neutrophils and to the development of severe
glomerulonephritis. Based on these, we have proposed that Mac-1 regulates
neutrophil activation in response to immune complexes. The SLE-associated R77H
variant of Mac-1 contributes to disease by allowing an amplified neutrophil
activation that leads to the instigation of disproportionate tissue injury (Rosetti et al, J Immunol 2012).

Role of Helios Transcription Factor as New Cancer-Associated Immunosuppressant and Strategies to Inhibit It
This project stems from two observations made by my working group: (1) CD8+ T cells become inactivated when they encounter their antigen expressed as self-antigen, and this inactivation involves the expression of the transcription factor Helios (IKZF2); (2) an analogous phenomenon is observed in tumor infiltrating CD8+ T cells. Based on this, we postulate the hypothesis that the induction of Helios in the tumor microenvironment slows down the immune system's ability to eliminate cancer cells. Therefore, the inhibition of Helios represents a novel therapeutic opportunity in the context of cancer. The objectives of this project are to test whether Helios limits the functional capacity of T cells, to identify the mechanism through which it does so, and to discover inhibitors of its induction. Link: https://www.youtube.com/watch?v=umx6mkhnzvM&ab_channel=LabInmunopatologia

La Oveja Eléctrica - Season 18 Trailer
Introducing at Dr. Jose Carlos CrispĂn Acuña in the program The Electric Sheep, the most prominent science dissemination program in Mexico and Latin America.