TEMAS
Papel de la fosfatasa PP2A en el control de la inflamaciĂłn
Estudios iniciales encontraron que la subunidad catalĂtica de la
fosfatasa de serinas y treoninas PP2A (PP2Ac) estĂĄ sobreexpresada en cĂŠlulas T
de pacientes con lupus eritematoso generalizado (LEG). Con el fin de investigar
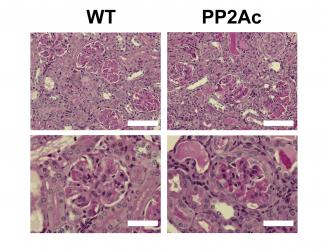
si dicho defecto contribuye al desarrollo de LEG, creamos un ratĂłn transgĂŠnico
que expresa niveles anormalmente altos de PP2Ac en cĂŠlulas T. El ratĂłn
transgĂŠnico demostrĂł tener mayor susceptibilidad al desarrollo de
glomĂŠrulonefritis inducida por complejos inmunes (CrispĂn et al., J Immunol 2012).
En estudios posteriores mostramos que el mecanismo molecular a travĂŠs del cual
el incremento en los niveles de PP2Ac genera susceptibilidad a la nefritis es
la facilitaciĂłn epigenĂŠtica de la transcripciĂłn de genes pro-inflamatorios como
la Il17a a travĂŠs de un mecanismo
dependiente de IRF4 (Apostolidis et al.,
J Biol Chem 2013). PP2A
tambiĂŠn juega un papel esencial en la biologĂa de las cĂŠlulas T reguladoras ya
que su deleciĂłn especĂfica en cĂŠlulas FoxP3+ anula su capacidad
supresora y causa una enfermedad autoinmune sistĂŠmica grave (Apostolidis et al., Nat Immunol 2016).
Mecanismos que limitan la duraciĂłn de la respuesta inmune
Con el fin de entender con mayor detalle la biologĂa de PP2A y su
contribuciĂłn al desarrollo de autoinmunidad, realizamos un estudio para
identificar quĂŠ subunidades reguladoras se expresan en cĂŠlulas T. Encontramos
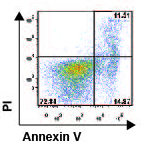
que B55Ă se induce en cĂŠlulas T durante estados de baja
concentraciĂłn de citocinas y que su presencia es necesaria y suficiente para la
muerte celular inducida por privaciĂłn de IL-2 (Crispin et al., Proc Natl Acad Sci
USA 2011). El anĂĄlisis de la cinĂŠtica de expresiĂłn de B55Ă en cĂŠlulas T de pacientes con LEG identificĂł a
un subgrupo de pacientes cuyas cĂŠlulas T son resistentes a la inducciĂłn de
apoptosis por privaciĂłn de IL-2. B55Ă no se
expresa en esos pacientes, lo que sugiere que fallas en la regulaciĂłn de esta
molĂŠcula podrĂan contribuir a la autoinmunidad al fallar la eliminaciĂłn de
clonas de cĂŠlulas T activadas.
DesapariciĂłn de la molĂŠcula CD8 como mecanismo de tolerancia perifĂŠrica
Los pacientes y los ratones con lupus frecuentemente tienen cantidades
anormalmente altas de una subpoblaciĂłn de cĂŠlulas T (TCR-ÎąĂ+) que carece de las molĂŠculas coreceptoras CD4 y CD8. Estas cĂŠlulas se
han denominado dobles negativas (DN). Nuestros estudios iniciales mostraron que
las cĂŠlulas T DN son capaces de producir grandes cantidades de citocinas
pro-inflamatorias (e.g. IL-17, IFN-gamma) y que son
abundantes en los infiltrados inflamatorios de riĂąones de pacientes con
nefritis lĂşpica (CrispĂn et al., J Immunol 2008). Estudios
posteriores encaminados a investigar el origen de estas cĂŠlulas encontraron que
provienen de cĂŠlulas CD8+ que pierden la expresiĂłn de CD8 tras la
activaciĂłn (CrispĂn et al., J Immunol 2009). Con el fin de
investigar con mayor detalle los factores que gobiernan la conversiĂłn de
cĂŠlulas CD8 a DN, establecimos un sistema in vivo usando ratones transgĂŠnicos.
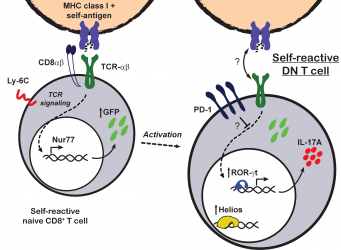
Demostramos que las cĂŠlulas CD8+ mantienen la expresiĂłn del
correceptor CD8 cuando son activadas en el contexto de una infecciĂłn. En
cambio, cuĂĄndo ĂŠstas se exponen a antĂgenos presentados como molĂŠculas propias,
sufren una inactivaciĂłn funcional que se mantiene por la desapariciĂłn de CD8 y
la expresiĂłn de niveles altos de molĂŠculas inhibitorias como PD-1 y Helios (RodrĂguez-RodrĂguez
et al., J Immunol 2015). Este proceso sucede en ratones normales en
forma continua ya que la subpoblaciĂłn de cĂŠlulas DN PD-1+ estĂĄ
formada por cĂŠlulas autoreactivas que solĂan ser CD8+ (RodrĂguez-RodrĂguez
et al., Eur J Immunol 2016).

Efectos biolĂłgicos de variantes genĂŠticas asociadas a enfermedades autoinmunes
Estudios de asociaciĂłn genĂŠtica (GWAS, genome-wide association studies), han detectado mĂşltiples genes que
confieren riesgo de desarrollar enfermedades complejas. Esos anĂĄlisis se basan
en la segregaciĂłn desequilibrada de polimorfismos comunes (SNPs, del inglĂŠs single nucleotide polymorphisms) entre
pacientes y personas sanas. Aproximadamente 30 loci han sido asociados a la
presencia de lupus. Una de las asociaciones mĂĄs fuertes se encuentra en el gen ITGAM, que codifica para la cadena alfa
de la molĂŠcula de adhesiĂłn Mac-1. El
alelo de ITGAM asociado a lupus provoca una mutaciĂłn (R77H). Para identificar
los efectos que causa la sustituciĂłn R77H, generamos lĂneas celulares que
expresan la variante normal y la variante de riesgo. En estudios in vitro
mostramos que la variante R77H disminuye la capacidad de Mac-1 de unirse a sus ligandos
(iC3b e ICAM-1), especĂficamente bajo flujo constante (Rosetti et al,
J Immunol 2012). En estudios
subsecuentes disecamos el mecanismo molecular que subyace al defecto: la
variante asociada a lupus (R77H) es incapaz de formar enlaces de captura:
âcatch bondsâ (Rosetti et al, Cell Reports 2015). Para estudiar los efectos in vivo de un
defecto funcional de Mac-1, utilizamos ratones genĂŠticamente modificados que
expresan receptores FcgRIIA humano en presencia o
ausencia de Mac-1. La inyecciĂłn intravenosa de suero de pacientes con LEG con
altas concentraciones de complejos inmunes, causan el depĂłsito de ĂŠstos en la
vasculatura glomerular. Este depĂłsito ocasionĂł el desarrollo de glomerulonefritis
Ăşnicamente en ratones deficientes de Mac-1. La ausencia de Mac-1 se asociĂł con un
gran infiltrado de neutrĂłfilos. Basados en esos trabajos, hemos propuesto que Mac-1
es una molĂŠcula clave que modula la respuesta de neutrĂłfilos a complejos
inmunes. Ausencia de Mac-1 o defectos en su funciĂłn se asocian a una mayor activaciĂłn
de neutrĂłfilos ante complejos inmunes y al desarrollo de daĂąo tisular (Rosetti et al, J Immunol 2012).

Papel del Factor de TranscripciĂłn Helios como un Nuevo Inmunosupresor Asociado a CĂĄncer y Estrategias para Inhibirlo
Este proyecto se deriva de dos
observaciones hechas por mi grupo de trabajo: (1) las cĂŠlulas T CD8+ se
inactivan cuando se enfrentan a su antĂgeno expresado como antĂgeno propio y
esa inactivaciĂłn involucra la expresiĂłn del factor de transcripciĂłn Helios
(IKZF2); (2) un fenĂłmeno anĂĄlogo se observa en las cĂŠlulas CD8 que infiltran
tumores malignos. En base a ello postulamos la hipĂłtesis de que la inducciĂłn de
Helios, en el microambiente tumoral, frena la capacidad del sistema
inmunolĂłgico de eliminar cĂŠlulas cancerosas. Por lo tanto, la inhibiciĂłn de
Helios representa una oportunidad terapĂŠutica novedosa en el contexto del
cĂĄncer. Los objetivos de este proyecto son probar si Helios limita la capacidad
funcional de las cĂŠlulas T, identificar el mecanismo a travĂŠs del que lo hace y
descubrir inhibidores de su inducciĂłn. Link: https://www.youtube.com/watch?v=umx6mkhnzvM&ab_channel=LabInmunopatologia

La Oveja ElĂŠctrica - Trailer Temporada 18
ParticipaciĂłn del doctor Jose Carlos CrispĂn AcuĂąa en el programa La oveja elĂŠctrica, el programa de difusiĂłn de las ciencias mĂĄs destacado en MĂŠxico y AmĂŠrica Latina.
Link: https://www.youtube.com/watch?v=Xio0eGjshFk&ab_channel=ConahcytM%C3%A9xico